Medical Device classification on the basis of Risk
Uncategorized Uncategorized
Medical Device classification on the basis of Risk
Health-care technology has become a critical component of health care, as it enables health- care providers to diagnose, treat, monitor and provide therapy to patients within an appropriate environment of care. Quality management of health-care technology helps ensure that these services are provided in a safe and effective way. The first step in managing health- care technology is to determine what items are to be managed and to create the health-care technology inventory. The inventory is a working document that is regularly checked and updated to accurately reflect the status of healthcare technology assets. When used appropriately, the inventory serves as an important and powerful tool to improve management of many key aspects of health-care technology. A medical device may be a surgical assisting application, or a mechanism of delivering drug into the human body. The purpose of using such a device is to provide specific medical assistance and long-term safety while minimising the device-related complications. The presentation of a systematic framework addresses the development of a medical device to meet health standards or safety limits by showing the integration of the required processes and instruments used in its design, evaluation, prototyping and delivery.
There has been a rapid introduction of computational modelling tools, medical imaging, optical-based flow measurements, sensor technology and manufacturing processes into the development of medical devices. The first stage of medical device development is design realisation and innovation. Design rules based on certain specifications defined along the process of realising the product can be achieved with reference to a set of product operation and safety guidelines.
Two types of design innovation exists:
1. Autonomous – design realisation independent from its other existing innovations, whereby a new design rule to increase the effectiveness of the device or its level of safety can be developed without redesign of the entire product.
2. Systemic – design realisation in conjunction with its related complementary innovations, whereby new design updates requires the need to reformulate the old design rules and redefine the design of the product.
Coordinating an autonomous or systemic design product innovation is particularly difficult when dealing with many design options. Therefore, computational modelling and design based on computer-aided design packages can help to lower product development cost significantly.
A medical devices is a product which is used for medical purpose in patient in diagnosis, therapy or surgery. The intended primary mode of action of a medical devices on the human body in contrast with that of medicinal products in metabolic, immunological, or pharmacological. Medical devices act by other means like physical, mechanical, thermal, physicochemical or chemical.
The term medical devices covers a vast range of equipment from simple wooden tongue depressor to most sophisticated implant or medical imaging system, haemodialysis machines. This means that medical devices are everything from bond aids to X-ray machines, contact lenses, hip implant, pacemaker, hospital beds and in vitro diagnostic devices.
Today medical devices are associated with almost all of medical problems dealt at the rural health clinic or in a large, specialised hospital. In other words, medical devices are now a pervasive part of modern medical care.
HISTORY
In 1906, the original Food and drug Act is passed by US Congress on June 30 and signed by president Theodore Roosevelt. It prohibits interstate commerce in misbranded and adulterated foods, drink and drug .this act did not cover medical devices.
In 1938 Federal Food, Drug and cosmetic Act is passed by congress, containing new provision for extending control to cosmetics and therapeutic devices.
In 1976 Medical Device Amendment passed to ensure safety and effectiveness of medical devices, including diagnostic products . The amendment require manufacturers to register with Food and Drug Administration (FDA) and follow quality control procedures.
The Amendment:
• defined a medical device,
• Established three devices classes( I,II,III)
• Identified pathway to market
• Established Advisory Panels, and
• outlined clinical investigation requirement
In 1990, Safe Medical Devices Act is passed, requiring nursing homes, hospitals, and other facilities that use medical devices to report to FDA incident that suggest that a medical devices probably caused or contributed to death, serious illness, or serious injury of a patient. Manufactures are required to control post- market surveillance on permanently implanted devices whose failure might cause serious harm or death.
In 1996, Facing House and Senate committee bill that would force it to streamline its procedures for drug and device approval, the FDA approves streamlined procedures only for AIDS drugs, cancer drug, and certain medical devices.
In 1997, Food and Drug Administration Modernization Act include measures to accelerate review of devices, regulate advertising of unapproved uses of approved drug and devices, and regulate health claims for foods.
In 2002, Medical Devices User Fee and Modernization Act passed, fees has been fixed for medical devices applications evaluation .provisions are establishment inspection by accredited third- parties.
In 2007, Food and Drug Administration Amendments Act represents a very significant addition to FDA authorit. Among the many components of the low ,the prescription Drug
User Fee Act (PDUFA) and the Medical Devices User Fee and Modernization Act the additional resources need to conduct the complex and comprehensive reviews necessary to new drug and devices.
The present chapter deals with introduction of medical devices, definition and their classification in different countries.
DEFINITION OF MEDICAL DEVICE
Medical Devices Definition According to GHTF
The Global Harmonization Task Force has proposed the following harmonized definition:‘ Medical device’ means any instrument, apparatus, implement, machine, appliance, implant, reagent for in vitro use, software, material or other similar or related article, intended by the manufacturer to be used, alone or in combination, for human beings, for one or more of the specific medical purpose of:
• Diagnosis, prevention, monitoring, treatment or alleviation of disease,
• Diagnosis, monitoring, treatment, alleviation of or compensation for an injury,
• Investigation, replacement, modification, or support of the anatomy or of a physiological process,
• Supporting or sustaining life,
• Control of conception,
• Disinfection of medical devices,
• Providing information by means of in vitro examination of specimens derived from the human body; and does not achieve its primary intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its intended function by such means.
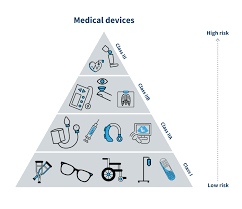
Classification of Medical Devices According to GHFT
Four risk classes of devices. The examples given are for illustration only and the manufacturer must apply the classification rules to each medical device according to its intended purpose.
Table 1.01: Classification of medical devices according to GHFT
CLASS RISK LEVEL DEVICE EXAMPLES
A Low Risk Surgical retractors , tongue depressors
B Low-moderate Risk Hypodermic Needles , suction equipment
C Moderate-high Risk Lung ventilator , bone fixation plate
D High Risk Heart valves , implantable
Definition of Medical devices According to WHO
According to WHO “Medical device means any instrument, apparatus, implement, machine, appliance, implant, reagent for in vitro use, software, material or other similar or related article, intended by the manufacturer to be used, alone or in combination, for human beings, for one or more of the specific medical purpose of:
1. Diagnosis, prevention, monitoring, treatment or alleviation of disease;
2. Diagnosis, monitoring, treatment, alleviation of or compensation for an injury;
3. Investigation, replacement, modification, or support of the anatomy or of a physiological process;
4. Supporting or sustaining life;
5. Control of conception;
6. Disinfection of medical devices;
7. Providing information by means of in vitro examination of specimens derived from the human body;
and does not achieve its primary intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its intended function by such means.
Medical Devices Definition in United States
According to US FDA, medical device is “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is:
1. Recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them,
2. Intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
3. Intended to affect the structure or any function of the body of man or other animals, and which does not achieve any of its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes.
Medical devices classification in US
US FDA has classified medical devices in three classes based on the degree of regulatory control necessary to ensure their safety and effectiveness show in table
Table 1.02 : Classification of medical devices in US
Classification Risk level Regulatory control Examples
Class I Low General control Adhesive bandages,
hospital beds, wheel chair
Class II Moderate Physician controlled
distribution, Moderate control Oxygen masks, Blood
pressure cuffs, sutures.
Class III Moderate
to High Need strict control for safety and efficacy purpose Pacemaker, Vascular grafts, Coronary stent.
Class I devices are those that present a low risk of harm to user and are subject to general control : See figure 2.01
Examples: Elastic bandages, Tongue depressors, Examination gloves, Hearing aids , Arm slings and Microbial analyzers.
Class II devices are more complicated and require special control for labelling, guidance, tracking, design, performance standards, and post market monitoring. Majority of these require Premarket Notification 510 (k) to get market entry in US
Examples : Powered wheelchairs, CT scanners, Contact lens care products and Infusion pump.
Class III devices usually sustain or support life, are implanted, or present potential unreasonable risk of illness or injury . They have the toughest regulatory controls. Most of these devices require Premarket Approval because general special control alone cannot reasonably assure their safety and effectiveness.
Examples: Pacemaker, Implanted weight loss devices, Non- invasive glucose testing devices, Medical imaging analyzers and Cochlear implants.
Medical Devices Definition in European Union
According to EU, “The New Approach, defined in a European Council Resolution of May 1985,(3) represents an innovative way of technical harmonisation. It aims to remove technical barriers to trade and dispel the consequent uncertainty for economic operators, to facilitate free movement of goods inside the EU.
The core legal framework consists of three directives:
1. Directive 90/385/EEC regarding active implantable medical devices
2. Directive 93/42/EEC regarding medical devices
3. Directive 98/79/EC regarding in vitro diagnostic medical devices
They aim at ensuring a high level of protection of human health and safety and the good functioning of the Single Market. These three main directives have been supplemented over time by several modifying and implementing directives, including the last technical revision brought about by Directive 2007/47 EC.
Directive 2007/47/EC defines a medical device as (paraphrasing): Any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, together with any accessories, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
1. Diagnosis, prevention, monitoring, treatment, or alleviation of disease;
2. Diagnosis, monitoring, treatment, alleviation of, or compensation for an injury or handicap;
3. Investigation, replacement, or modification of the anatomy or of a physiological process;
4. Control of conception;
This includes devices that do not achieve their principal intended action in or on the human body by pharmacological, immunological, or metabolic means—but may be assisted in their function by such means.
Medical devices classification in European Union
In EU medical devices are classified according to their intended use .there are 18 rules for classification of medical devices . These rules are applied to help a manufacturer determine

Related Posts
PRESERVING HISTORY: THE ART AND SCIENCE OF RESTORING HERITAGE BUILDINGS
PRESERVING HISTORY: THE ART AND SCIENCE OF RESTORING HERITAGE BUILDINGS Introduction: Heritage buildings serve as tangible embodiments of the past, carrying within their walls the intricate narratives of history, culture, and architectural evolution. From ancient temples to medieval castles, from
Cat-Scratch Disease: An approach to diagnosis and treatment
Mr. Vimal Pathak M.Pharm 2nd Sem GIP (Geeta University, Naultha Panipat ) Introduction: A cat’s bite or scratch can transmit the infectious disease known as cat-scratch disease (CSD). When someone bites or scratches someone with cat scratch illness, a non-painful

Dining Etiquette and Its Importance
Dining Etiquette and Its Importance: Dining etiquette, often overlooked in our fast-paced and modern world, plays a crucial role in our social interactions, personal relationships, and professional life. The way we conduct ourselves at the dining table reflects our upbringing,